Segawa-Syndrom als Dopaminmangelerkrankung
Das Segawa-Syndrom gehört in das große Krankheitsbild der Dystonien. Als Dystonie bezeichnet man Bewegungsstörungen, die durch unwillkürliche, langsame, anhaltende Muskelkontraktionen charakterisiert sind. Diese führen zu drehenden, sich wiederholenden Bewegungen oder abnormen Haltungen. Die einzige, kausal behandelbare Dystonie ist das Segawa-Syndrom, welches leider oft sogar von Neurologen mit Parkinson verwechselt wird.
Entdeckungsgeschichte des Segawa-Syndroms
Das Segawa-Syndrom und die Dystonien sind innerhalb der Neurologie noch relativ junge Krankheitsbilder. Der deutsche Neurologe Oppenheim führte 1911 den Begriff Dystonie ein, als er ein Syndrom mit Gangstörung als Dystonia musculorum deformans bezeichnete. Jedoch schon 1908 wurde dieses Krankheitsbild von seinem Fachkollegen Schwalbe als „eigentümliche tonische Krampfform mit hysterischen Symptomen“ beschrieben (Ceballos-Baumann, 1996; S. 89). Hinzugekommen sind viele neue Erkrankungsformen, die nicht nur Ähnlichkeiten in der Symptomatik, sondern auch Verwandtschaft in der pathophysiologischen Grundstruktur aufweisen. Eine dieser Erkrankungen ist das Segawa-Syndrom, eine seltene Dystonie, die der japanische Neurologe Masaya Segawa erstmals 1970 als eigenes Krankheitsbild beschrieb und somit Namensgeber dieses Krankheitsbildes wurde. Im Jahre 1976 erschien der erste englischsprachige Bericht (Segawa et al. 1976). In Fachkreisen ist das Segawa-Syndrom auch als Dopamin-Responsive-Dystonie (DRD) bekannt. Heute wird sie häufig als DYT5-Dystonie bezeichnet.
Klinik und Symptome der Erkrankung
Als früheste Symptome treten in der ersten Lebensdekade dystone Haltungen, zum Beispiel die Einwärtsdrehung der Füße, auf. Die Erkrankung verläuft unbehandelt in der Regel fortschreitend und erfasst die gesamte untere Extremität. In schweren Fällen generalisiert die Erkrankung und führt zu völliger Bewegungsunfähigkeit. Außer Dystonie finden sich beim Segawa-Syndrom eines oder mehrere Symptome der Parkinson-Erkrankung wie Rigor (erhöhte Steifigkeit der Muskeln), Bradykinese (Bewegungsverarmung und -verlangsamung), Tremor (Zittern), Dyskinesie (plötzlich auftretende Fehlbewegungen und Verkrampfungen der Muskulatur) und der Verlust von Haltungsreflexen. Die Ausprägung der Symptome unterliegt häufig einer tageszeitlichen Schwankung. In etwa 75 % der Fälle ist die Symptomatik in den Morgenstunden oder nach einer längeren Schlafphase gebessert und verschlechtert sich dann im Verlauf des Tages.
Diagnose
Für einen mit dem Krankheitsbild vertrauten Neurologen ist die Diagnose extrem einfach, da der Verdacht des Segawa-Syndroms oft schon durch eine gezielte Befragung des Patienten bestätigt werden kann. Die Dystonie ist ein fehlregulierter Spannungszustand bestimmter Muskelgruppen, der durch die unwillkürlichen Verkrampfungen zu bizarren Körperhaltungen und Bewegungen führen kann. Im speziellen Fall des Segawa-Syndroms ist eine ausführliche Befragung von Eltern und betroffenem Kind wegweisend. Kein Labortest oder funktioneller Nerventest gibt einen spezifischen Hinweis auf die Erkrankung. Die Untersuchung des Patienten zeigt die für die Dystonie typischen Muskelverkrampfungen und das Muskelzittern. Der Arzt muss in dieser Situation nachfragen, ob nach dem Schlafen oder nach längeren Ruhephasen die Symptome weniger stark ausgeprägt sind und nach stärkeren körperlichen Belastungen (z.B. Spiel mit Freunden oder spazieren gehen) in ihrer Ausprägung deutlich zunehmen. Kinder können am Morgen oft selbst essen und müssen am Abend gefüttert werden. Bei dieser Konstellation von Symptomen und Krankengeschichte ist der Goldstandard der Diagnosestellung eine Testdosis von Dopamin zu verabreichen. Die Wirkung von Dopamin setzt innerhalb von Stunden ein und beseitigt fast vollständig alle Symptome des Segawa-Syndroms.
Fehldiagnosen
Beim Segawa-Syndrom handelt es sich um ein sehr seltenes und deshalb leider bei den Ärzten wenig bekanntes Krankheitsbild. Da die Symptome bei der Geburt kaum oder gar nicht ausgeprägt sind und keine perinatale Diagnostik möglich ist, wird die Diagnosestellung oft über Jahre verschleppt. Sollten Symptome schon bei einem Säugling auffallen, wird in der Regel ein verzögerter Geburtsverlauf mit einer Sauerstoffschädigung des Mittelhirns verantwortlich gemacht. Den betroffenen Eltern wird dann oft empfohlen, intensive Krankengymnastik zu betreiben, da möglicherweise ein geburtsbedingter Sauerstoffschaden des Gehirns zu irreversiblen Schäden geführt hat, die man durch physiotherapeutische Übungen in seiner Wirkung abschwächen kann. Leichte Symptome nach dem ersten Lebenshalbjahr fallen manchmal einem aufmerksamen Kinderarzt auf, der eine Bewegungsarmut der Beine, einen verzögerten Laufbeginn oder die Einwärtsdrehung meist des rechten Fußes feststellt. Es beginnt dann nicht selten für Eltern und Kind eine lange Odyssee durch verschiedene Fachgebiete der Medizin. Weil die Einwärtsdrehung des Vorfußes das häufigste Frühsymptom ist, wird der Kinderorthopäde als Erstes aufgesucht. Da die Dystonie normalerweise kein typisch orthopädisches Krankheitsbild verursacht und weitere hinweisgebende Symptome fehlen oder noch nicht auffallen, ist die häufigste Fehldiagnose eine Schwäche des sogenannten Peroneusnerven. Meist wird deshalb zunächst eine konservative Therapie beschritten, bei der durch Anlegen einer orthopädischen „Thomasschiene“, besonders in der Nacht, der Spitzfußstellung entgegengewirkt werden soll. Da aufgrund der Fehldiagnose naturgemäß keine Besserung eintreten kann, wird von orthopädischer Seite oft eine Verlängerungsoperation der Achillessehne oder eine andere Sehnenumstellungsoperation des Vorfußes vorgenommen, d.h. im ungünstigsten Fall wird die Diagnose über Monate und Jahre verschleppt und schließlich als Verzweiflungstat eine eingreifende, medizinisch nicht indizierte, verstümmelnde orthopädische Operation gewählt. Die vermutete Schwäche des Peroneusnerven ruft oft den Neurologen auf den Plan. Da rein statistisch ein normaler Neurologe während seiner Lebensarbeitszeit nur einmal die Gelegenheit hat, die Erstdiagnose eines Segawa-Syndroms zu stellen, besteht leider auch hier eine große Wahrscheinlichkeit, dass die Diagnose nicht auf Anhieb korrekt gestellt wird. Der Patient durchwandert meistens zunächst einen Irrgarten von neurologischen Untersuchungsmethoden. Der Besuch beim Neurologen findet naturgemäß oft in den Vormittagsstunden statt, dann, wenn die Symptome am wenigsten ausgeprägt sind. Bei vielen Patienten mit Segawa-Syndrom wird die Nervenleitungsgeschwindigkeit des Peroneusnerven unter Stimulation als völlig normal gemessen. Eine Nerven- und Muskelbiopsie erbringt häufig auch eine völlig normale Gewebestruktur. Oft wird auch noch eine Nervenentzündung in Betracht gezogen, die zu einer Punktion des Gehirnwassers (Lumbalpunktion) führt. Größeres Glück mit einer früheren Diagnosestellung können Patienten haben, die glücklicherweise in den Abendstunden untersucht werden, nämlich dann, wenn zusätzliche Symptome wie Muskelkrämpfe oder Muskelzittern zur allgemeinen Muskelsteifheit hinzukommen. Überhaupt sorgt das oft tageszeitlich wechselnde Krankheitsbild zur Verwirrung von Eltern und Ärzten, da sich morgendliches normales Verhalten (aufgefüllte Dopaminspeicher) mit Muskelkrämpfen und Zittern nach starken körperlichen Belastungen (entleerte Dopaminspeicher) in bunter Folge abwechseln. Nicht wenige Patienten landen deswegen in der Psychiatrie, da ihnen Simulation, vegetative Dystonie oder versteckte Depression unterstellt werden. Die voreilige Verordnung von Tranquillizern (Beruhigungsmitteln) führt leider zu einer Abmilderung der typischen Leitsymptome der Erkrankung und oft zu einer fatalen Verschleierung der typischen Symptome mit psychiatrischen Fehldiagnosen, die alleine durch die medikamentöse Therapie zu erklären wären. Ein verschwindend kleiner Teil der Patienten wird aufgrund des in den Abendstunden vorherrschenden Zitterns als atypischer Parkinson eingestuft: nur sie erhalten glücklicherweise das typische Parkinsonmittel L-Dopa und gehen damit einer schnellen Behandlung entgegen. Sie werden mit dem einzig richtigen Medikament behandelt, allerdings ohne dass sie jemals ihre wahre Diagnose erfahren. Da das Segawa-Syndrom auch Verspannungen im Bereich der Wirbelsäule erzeugen kann, die unbehandelt zu Fehlstellungen der Wirbelkörper führen, verbleibt über 1/3 der Patienten in orthopädischer Behandlung, ohne der Diagnose einen Schritt näher zu kommen.
Therapiemöglichkeiten unter dem Aspekt der Dosierung
Sollte die Diagnose Segawa-Syndrom gestellt worden sein, so ist die Therapie einfach. In den meisten Fällen mindert eine Dosis von 20 mg pro kg Körpergewicht pro Tag von L-DOPA die Symptome vollständig. Da L-DOPA auch zur Parkinsonbehandlung und zum Eindämmen des „Restless-Legs-Syndrom“ (Ruhelosigkeit der Beine) verwendet wird, ist es leicht und weltweit erhältlich. Es ist auch eine vergleichsweise billige Therapie. Die Strukturformel von L-DOPA ist:
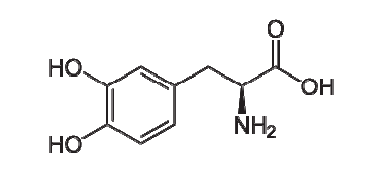
Abbildung 9: Strukturformel von L-DOPA
Als Arzneistoff wurde L-DOPA unter dem Namen Madopar® 1973 von Hoffmann-La Roche erstmals auf den Markt gebracht. Heute gibt es verschiedene Präparate, wobei L-DOPA in Kombination mit verschiedenen Decarboxylasehemmern verwendet wird:
- Madopar® (L-DOPA, Benserazid); Roche
- Nacom® (L-DOPA, Carbidopa); Bristol-Myers Squibb GmbH
Warum wird L-DOPA und nicht Dopamin verabreicht? Der eigentliche Wirkstoff Dopamin kann die oben beschriebene Blut-Hirn-Schranke nicht überwinden. L-DOPA wird als „Prodrug“ bezeichnet. Es handelt sich dabei um die Vorstufe des eigentlichen Wirkstoffs. Die medikamentös wirksame Form Dopamin wird aus L-DOPA unter CO2-Abspaltung im Körper gewonnen.
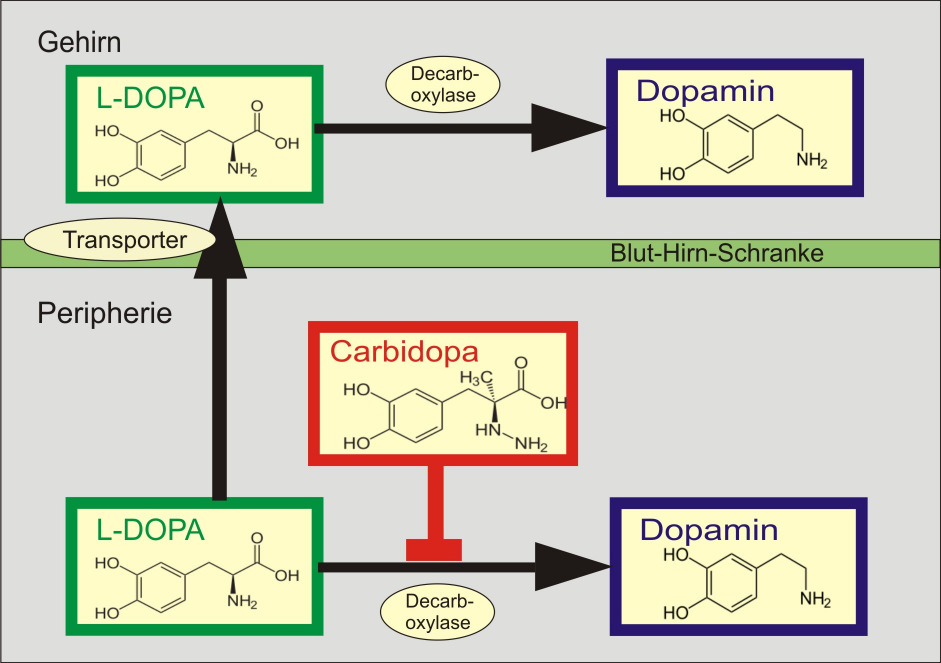
Abbildung 10: Wirkmechanismus eines Kombinationspräparates aus L-DOPA und Carbidopa (nach www.wikipedia.org/wiki/Parkinsonmittel 11.3.2008)
L-DOPA aber muss mit Decarboxylase-Hemmern kombiniert werden, um eine Abspaltung von CO2 von L-DOPA vor dem Passieren der Blut-Hirn-Schranke zu vermeiden. Ohne Decarboxylierungshemmung würde 95 % des verabreichten L-DOPAs bereits außerhalb des Gehirns decarboxyliert und damit unwirksam gemacht werden. Der Decarboxylase-Hemmer wiederum kann die Blut-Hirn-Schranke nicht passieren, was eine CO2-Abspaltung im Gehirn zur Folge hat. Somit liegt nun der gewünschte Wirkstoff Dopamin im Gehirn vor.
Die tatsächliche Dosierung des Medikaments muss durch langsames Herantasten an die optimale Verabreichungsmenge für jeden Patienten individuell herausgefunden werden. Ist die Dosierung zu niedrig, so treten die Symptome des Segawa-Syndroms noch in verminderter Form auf. Die Muskelkrämpfe werden zwar schwächer und das Nachinnendrehen der Füße ist weniger stark ausgeprägt, jedoch noch deutlich erkennbar. Sollte zu viel des Medikamentes verabreicht werden, so kommt es zu Hyperaktivität, zittrigen und fahrigen Bewegungen, welche ruhiges Stillsitzen unmöglich machen. Ist die richtige Dosierung für den Patienten gefunden worden, so ist dieser völlig symptomfrei. Bei Kindern, die sich noch im Wachstum oder in der Pupertät befinden, muss das Medikament immer wieder neu dosiert werden und dem Köperwachstum angepasst werden. Es ist von Vorteil, die Tagesdosis auf mehrere Einnahmen zu verteilen. Zum Beispiel morgens zum Frühstück eine Einheit einnehmen und eine zweite Einheit zum Mittagessen, welche dann bis zum Abend wirkt. Sollte der Patient Abends nach einem anstrengenden Tag ein Zittern verspüren, ist es vorteilhaft eine Tablette einzunehmen, um besser einschlafen zu können.
Nebenbemerkungen zu L-DOPA:
- Im Buch „Awakenings“ (1973, Vintage Books) von Oliver Sacks schildert der Neurologe sehr anschaulich das Herantasten an die optimale Dosierung von L-DOPA und die leider nur kurzzeitigen Therapieerfolge bei Patienten, welche an Europäischer Schlafkrankheit (Encephalitis lethargica), einer Gehirnentzündung, erkrankt sind.
- Der Nobelpreis für Medizin wurde 2000 an den schwedischen Wissenschaftler Arvid Carlsson vergeben, der als erster Forscher Mäusen mit Parkinsonsymptomen L-DOPA verabreichte und somit die Wirksamkeit des Medikaments L-DOPA im Tierexperiment nachwies.
- Der Nobelpreis für Chemie ging 2001 an William S. Knowles für seine Arbeit an der Synthese von L-DOPA.
Genetik
Das Segawa-Syndrom wird normalerweise autosomal-dominant vererbt, wobei Frauen weitaus häufiger betroffen sind als Männer (4:1). Bei den meisten Segawa-Betroffenen finden sich keine weiteren Familienangehörigen, die auch betroffen sind. Dies wurde damit erklärt, dass ein Großteil der Segawafälle auf Neumutationen zurückzuführen ist oder, dass aufgrund der unvollständigen Penetranz des Merkmals nur sehr wenige Träger des mutierten Gens auch erkranken. Verschiedene Gendefekte können zum gleichen Krankheitsbild führen.
Das Segawa-Syndrom ist somit eine heterogene Erkrankung:
- Es fand sich ein Gen auf Chromosom 14, welches für das Enzym GTP-Cyclohydrolase I (GCH I) codiert (siehe Synthese von Dopamin). Durch molekulargenetische Untersuchungen konnten Mutationen in dem Gen für GCH I bei Segawa-Patienten nachgewiesen werden. Diese Mutationen führen zu einer Verminderung der Enzymaktivität und damit zu einer verringerten Dopamin-Synthese. Die GTP Cyclohydrolase I wird durch das Gen GCH I auf Chromosom 14q22.1-q22.2 kodiert. Das Gen besteht aus 6 Exonen, die über 30 kb verteilt sind. Durch Mutationen in diesem Gen entsteht das Krankheitsbild des Segawa-Syndroms.
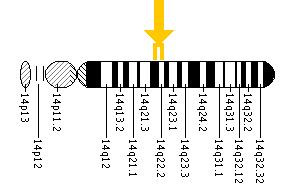
Abbildung 11: Chromosom 14 mit GCH I-Genlocus (aus http://ghr.nlm.nih.gov/gene=gch1 11.03.2008)
- Es ist auch eine vermutlich autosomal-rezessiv vererbte Form des Segawa-Syndroms im Gespräch. Bei dieser handelt es sich um eine Mutation auf Chromosom 11 (Gen besteht aus 14 Exonen), wobei die Tyrosinhydroxylase nur unzureichend gebildet werden kann.
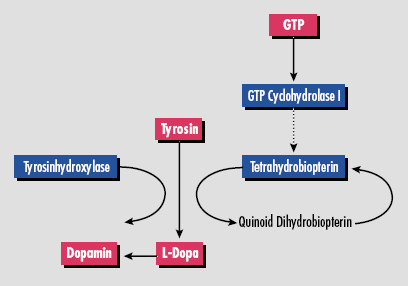
Abbildung 12: Wirkstellen von GTP I und Tyrosinhydroxylase (nach Deutsches Ärzteblatt 93, Heft 24, 14.Juni 1996 (57))
| Modus der Vererbung |
Chromosomale Lokalisation |
Mutation im Gen codierend für |
| autosomal-dominant |
14q22.1-q22.2 |
GTP-Cyclohydrolase I |
| autosomal-rezessiv |
11p15.5 |
Tyrosinhydroxylase |
Tabelle 3: Enzyme und korrespondierende Gene
Epidemiologie, Verbreitung
Das Segawa-Syndrom ist eine seltene Krankheit mit geschätztem Vorkommen von 0,5 bis 1 pro 1 Mio. Einwohner, d.h. in Deutschland gibt es nur knapp 100 Patienten. Von den meisten Fällen wird in Japan und Südostasien berichtet. Aber mit steigendem Bewusstsein für diese Krankheit wird man auch in anderen Teilen der Welt mehr Erkrankungen entdecken. Epidemiologische Studien über das Auftreten des Segawa-Syndroms fehlen.
Schizophrenie
Dieses Kapitel können Sie in der
Facharbeit nachlesen.